5.0 Mesh Generator¶
In order to build a MINEDW model, the user must first create a 2-D mesh using either the Grasshopper plugin for Rhinoceros 3D (Rhino, a 3-D modeling program) that is distributed with MINEDW or any other software designed to create a 2-D mesh. The 2-D mesh needs to be saved as either a stereolithography (.STL) file or a MINEDW geometry (.PST) file. The 2-D mesh file can then be imported to the main MINEDW GUI where the rest of the model setup occurs.
5.1 Creating a Mesh¶
For the creation of a mesh, Rhino should be installed. After the successful installation of Rhino, copy the “MMesh.gha” and “triangulation.dll” files that were provided in the “bin” directory of the installed directory of MINEDW into the directory “C:\Users\[YOUR USER NAME]\Application Data\Grasshopper\Libraries.”
Open Rhino and import or create any features you would like to include in your mesh. This can be done using the Rhino drawing tools or by importing or copying from AutoCAD .DXF files. Launch Grasshopper by typing “Grasshopper” into the Rhino command line, or double-click the “Launch Grasshopper” tool icon to open the Grasshopper window.
From the Grasshopper window, open the “template.gh” file that is in the “bin” directory of the installed MINEDW directory. A workflow will appear on the Grasshopper canvas, shown in Figure 5.1. This workflow is designed to create a 2D mesh for a typical mining-related groundwater flow model. The workflow may be modified using the tools contained in Rhino and Grasshopper to suit any requirements.
Rhino and Grasshopper are two powerful external visualization and mesh-generation tools that are not developed by Itasca. Users are strongly encouraged to read the operational manuals of Rhino and Grasshopper to become familiar with these tools.
The user is also recommended to work through tutorials for mesh generation using Rhino and Grasshopper.
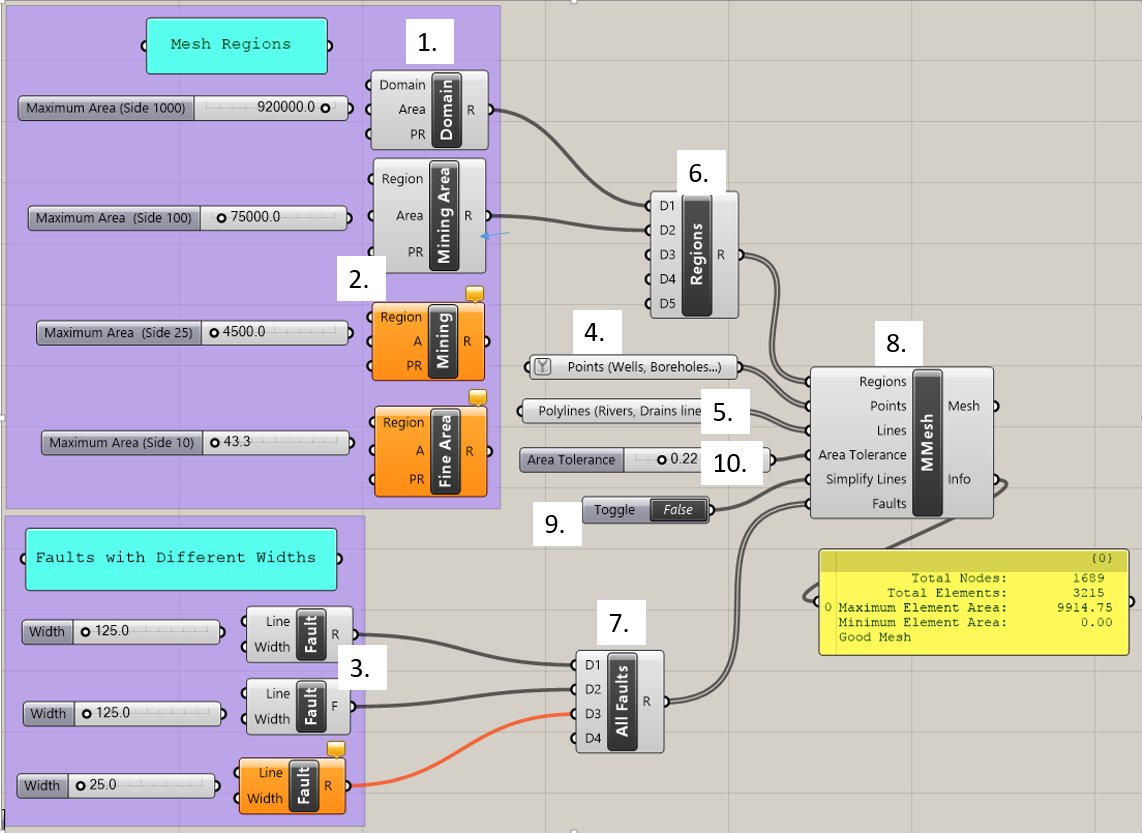
Figure 5.1 The Grasshopper mesh generator template¶
The blocks shown in Figure 5.1 are called components. Each of the key components is labeled by number and will be explained in detail in Section 5.2. Components have input parameters on their left side and output parameters on their right side. A component box is orange in color if it has a warning message. The message can be viewed in the comment box on top of the component. The user assigns features such as rivers, wells, and faults to components in Grasshopper to add features to the mesh. The grey “All Faults” and “Regions” (6 & 7) components are used to combine multiple inputs of the same type. The grey slider blocks, if connected to another component’s input parameter, allow the user to adjust parameters such as maximum element area and fault width. The “MMesh” (8) component creates a mesh that incorporates all the components it is connected to. The yellow box below the “MMesh” component gives information about the mesh, including the number of vertices and elements and the minimum element size once it is created. Components may be connected by clicking on the half circle next to an input parameter and dragging the mouse to the half circle next to an output parameter. Components may be disconnected by right-clicking on a parameter at either end of the connection and choosing the “Disconnect” option. Once the components have been connected and assigned with the proper construction features, the mesh can be viewed and will dynamically update as changes are made to various components. The components and the steps involved in making a mesh are discussed in detail in the following sections.
5.2 The Mesh Components¶
The template contains the following components, which are labeled by number in Figure 5.1:
Domain: This component is used to define the extent of the model domain. The input will be a single, closed polyline curve.
Mining Area, Mining, and Fine Area: These components are used to outline regions of the model domain that will have a finer mesh discretization. The input will be one or more closed polyline curves.
Fault: This component is used to define any faults in the model domain. The mesh generator will ensure that model nodes follow the line of the fault and will apply a width of the fault for a more accurate geologic representation. The user may specify the width of a fault. The input for this feature is a curve that represents the centerline of the fault.
Points: This component is used to specify any points that need to correspond to nodes in the mesh, such as a pumping well. The input for this feature is one or more points.
Polylines: This component is used to specify any curves that need to correspond to edges in the mesh, such as rivers or streams. There is no width associated with this feature. The input for this feature is one or more curves.
Regions: This component combines all the mesh regions created in the “Mesh Regions” group. The output of this component is connected to the regions input parameter of the “MMesh” component.
All Faults: This component combines all the faults created in the “Faults with Different Widths” group. The output of this component is connected to the faults input parameter of the “MMesh” component.
MMesh: This component receives input parameters from the regions, faults, lines, and points that the user specifies and then generates a mesh. Information about the mesh appears in the yellow box.
Toggle: This component toggles between “True” and “False” values. The output of this component is connected to the “Simplify Lines” parameter: If the value is “True,” “MMesh” will simplify line inputs into the mesh; if the value is false, “MMesh” does not simplify line inputs.
Area Tolerance: This component specifies the minimum ratio between the actual area of an element and the maximum area assigned to the mesh region it belongs to.
The user must import or create each feature in Rhino prior to connecting the features to the Grasshopper workflow. Note that all features must be located in the same 2-D plane. To connect features that exist in the Rhino workbook to the Grasshopper workflow, right-click on the input parameter in the top left corner of the component block and choose “Set One Curve/Point” or “Set Multiple Curves/Points.” Then select the desired feature in Rhino. This process is described in detail for each type of component in the following sections.
For the “Mesh Regions” components, in the purple box in the upper left of the Grasshopper worksheet, all active components must be connected to the “Regions” component. The “Regions” component must then be connected to “MMesh” at the terminal labeled “Regions.”
5.2.1 The “Domain” Component¶
The “Domain” component (Figure 5.2) is used to specify the model domain boundary. All parts of the mesh must be contained within the model domain boundary. The input for this component must be a closed polyline curve. Note that curves that the user draws in Rhino may be non-uniform rational basis spline (NURBS) curves. NURBS curves cannot be used in the Grasshopper workflow but may be easily converted to polylines using the “Convert” command in the Rhino command line. See the Rhino documentation for more information. To connect a closed curve in the Rhino workbook to the “Domain” component in Grasshopper, right-click on the “Domain” input parameter in the upper left of the component (Figure 5.2a). Select “Set One Curve” (Figure 5.2b). Then select the curve that delineates the model domain boundary in the Rhinoceros window.
The user can check that the desired curve was connected to the “Domain” component by clicking on the “Domain” component so that it is highlighted in green. In the Rhino workbook, any curve that is assigned to the highlighted component will turn green. Curves may be disconnected from the “Domain” component by right-clicking on the “Domain” input parameter and selecting the “Clear Values” option in the “Domain” menu, shown in Figure 5.2b.
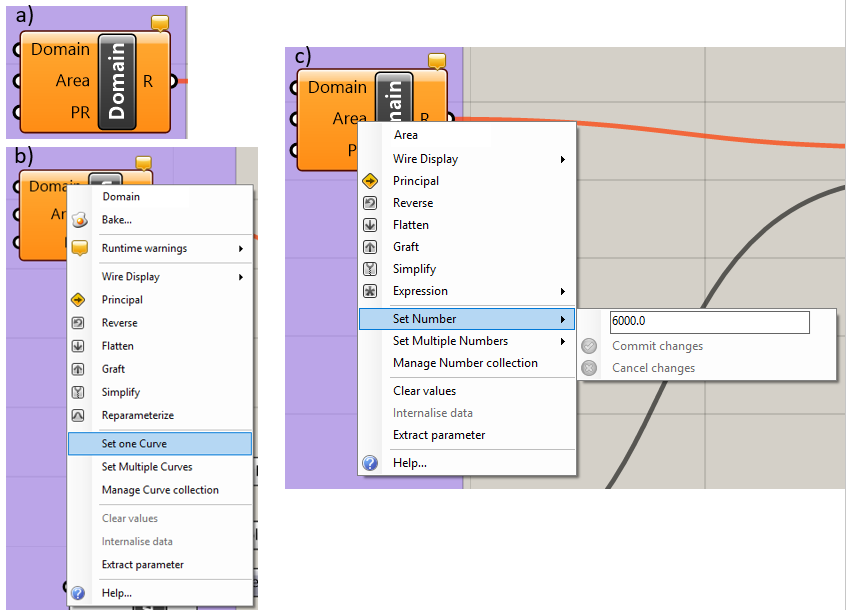
Figure 5.2 The “Domain” component: a) the component, b) the “Domain” input parameter menu, c) the “Area” input parameter menu¶
The maximum element area is set in two ways depending on whether the “Maximum Area” block and “Area” link in the “Domain” block are connected or not.
If the “Maximum Area” block and “Area” link in the “Domain” block are not connected, the user can right-click on the “Area” input parameter, which brings up the menu shown in Figure 5.2c. Click on the “Set Number” menu option and enter the maximum element size desired for the mesh; note that the user will have the ability to add regions of finer discretization.
If the “Maximum Area” block and “Area” link in the “Domain” block are connected, it also is only possible to set the maximum element area by connecting the grey slider to the left of the component, shown in Figure 5.3. The user can either use the slider to determine the value of the maximum area or double-click the grey area and enter the value.
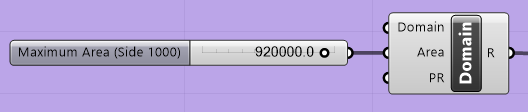
Figure 5.3 The “Maximum Area” slider¶
It should be noted that if the “Maximum Area” block is connected to the “Area” link of the “Domain” block, the value in the “Maximum Area” block will supersede the value that is entered in the “Area” parameter in the “Domain” block.
Blocks may be connected by clicking on the half circle of the “Maximum Area” block and dragging the mouse to the half circle of the “Area” parameter in the “Domain” block. Blocks maybe disconnected by right-clicking the specific parameter in the “Domain” block (i.e., “Area” in Figure 5.3) and selecting “Disconnect” from the pop-up menu.
5.2.2 The “Mining Area,” “Mining,” and “Fine Area” Components¶
The “Mining Area,” “Mining,” and “Fine Area” components are intended to allow the user to assign regions of increased mesh density to the area surrounding the mining activities, the location of the mining activities themselves, and any other areas that require it. The regions that the user assigns to any of these three components must be contained within the model domain that the user specified in the “Domain” component. These regions of fine mesh discretization may be located completely inside or completely outside of another fine mesh discretization region; however, they cannot partially overlap.
The “Mining Area,” “Mining,” and “Fine Area” components are used in the same way as the “Domain” component. The user right-clicks on the “Region” input parameter and selects the menu option “Set One Curve” or “Set Multiple Curves” (see Figure 5.2b). The user then selects the curve or curves that delineate the region that requires a finer mesh. The user may specify the maximum element area in the region by right-clicking on the “Area” input parameter and selecting “Set Number” from the menu. Alternatively, the user may set the maximum element size by connecting the “Area” input to the “Maximum Area” slider and using the slider to select the desired maximum element size, as shown in Figure 5.3, or enter the value as described in Section 5.2.1.
5.2.3 The “Fault” Component¶
The “Fault” component (Figure 5.4a) is used to create faults that have width. The user must first create or import a curve that delineates the center line of a fault in Rhino. The user right-clicks on the “Line” input parameter and selects the menu option “Select One Curve,” shown in Figure 5.4b. The user then selects the desired curve in Rhino. The user may also select the “Select Multiple Curves” option and then select all the faults that have the same width. If different widths are desired, the user must use multiple “Fault” components. To disconnect curves, select the “Clear Values” option in the “Line” input parameter menu, shown in Figure 5.4b.
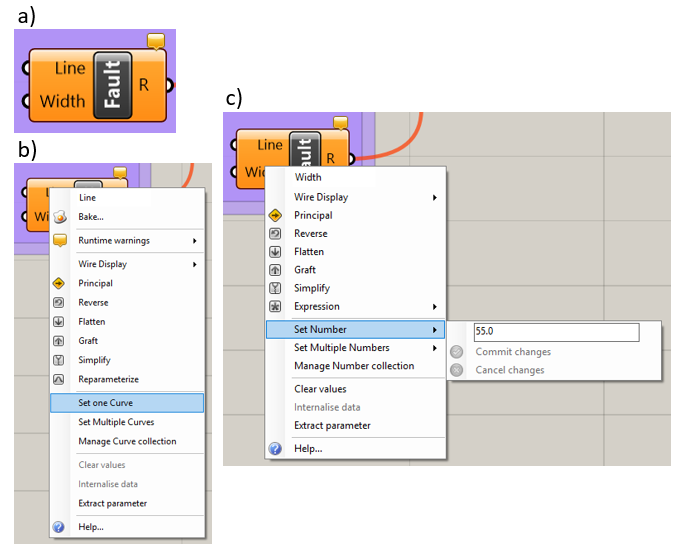
Figure 5.4 The “Fault” component: a) the component, b) the “Line” input parameter menu, c) the “Width” input parameter menu¶
The width of the fault is specified for each “Fault” component by right-clicking on the “Width” input parameter and selecting the menu option “Set Number,” then entering the desired fault width. This is shown in Figure 5.4c. Alternatively, the user may connect the “Width” slider, shown in Figure 5.5, to the “Width” input parameter and then use the slider to specify the desired fault width. Note that if the slider is connected, the “Set Number” option will be greyed out.
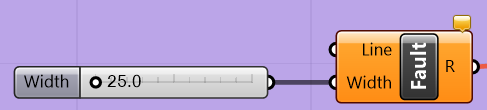
Figure 5.5 The “Fault” component with the width parameter slider¶
5.2.4 The “Points” Component¶
The “Points” component (Figure 5.6a) allows the user to specify points that will correspond to vertices in the mesh. This is typically used for pumping wells or monitoring wells so that the code’s calculation point will exactly match the location of the point feature. To specify hard points, the user right-clicks on the “Points” component and then selects either “Set One Point” or “Set Multiple Points,” shown in Figure 5.6. The user then selects the desired point or points in the Rhino workbook. The “SelPt” command may be used to select all points in the Rhino workbook. The command line will say “Point object to reference (Type=Point),” where the type is “Coordinate” or “Point.” If the type is “Coordinate,” Grasshopper will save the coordinates of the points at the time it is connected to the Grasshopper workflow. If the point is moved later, Grasshopper will not update its location. When the “Point” type is used, Grasshopper will update the mesh if the point is moved later.
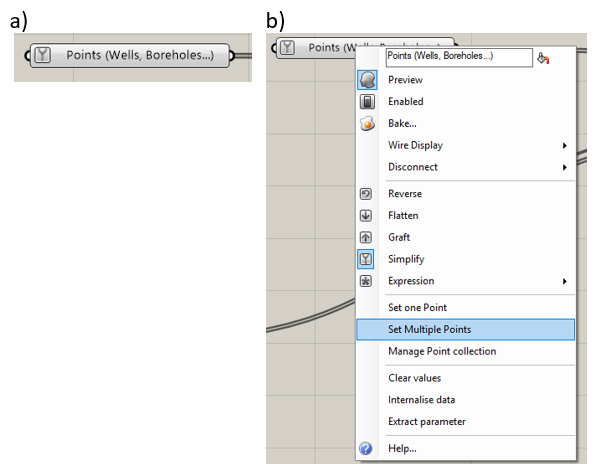
Figure 5.6 The “Points” component: a) the component, b) menu options¶
5.2.5 The “Polylines” Component¶
Curves assigned to the “Polylines” component will correspond to mesh edges. This is typically used for rivers or other features that are represented as curves and do not have a width assigned to them. The component is shown in Figure 5.7a. To specify a hard curve, right-click on the “Polylines” component and select the “Set One Curve” or “Set Multiple Curves” option, as in Figure 5.7b. Select the curve or curves that delineate the desired hard curves.
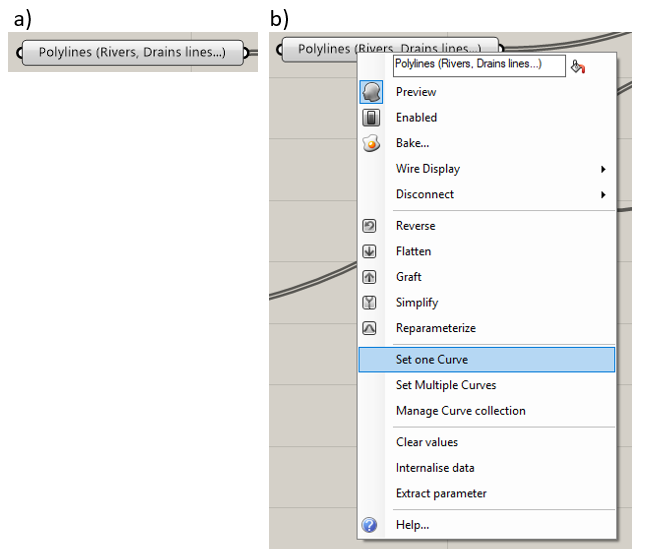
Figure 5.7 The “Polylines” component and menu options¶
5.2.6 The “Regions” Component¶
The “Regions” component receives the outputs from each component in the “Mesh Regions” group, merges them together, and outputs the result to the “MMesh” component. The component is shown in Figure 5.8.
Figure 5.8 The “Regions” component¶
5.2.7 The “All Faults” Component¶
The “All Faults” component receives the outputs from each component in the “Faults with Different Widths” group, merges them together, and outputs the result to the “MMesh” component. The component is shown in Figure 5.9.
Figure 5.9 The “All Faults” component¶
5.2.8 The “MMesh” Component¶
The “MMesh” component, shown in Figure 5.10, receives outputs from the “Regions,” “Points,” “Polylines,” and “Faults” components discussed above. In addition, “MMesh” has an “Area Tolerance” input parameter, which is the ratio of the smallest permissible area to the corresponding maximum area constraint. There is also a “Simplify Lines” input parameter, which receives a Boolean value from the “Toggle” component. If the value is “True,” all polyline inputs will be simplified, meaning the number of control points will be reduced and redistributed. This is recommended if there is a small angle between curve features. Both the “Area Tolerance” and “Toggle” components are discussed in further detail in the following sections.
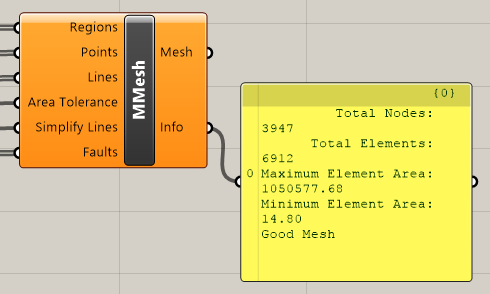
Figure 5.10 The “MMesh” component¶
5.2.9 The “Toggle” Component¶
The user assigns either a “True” or a “False” value to the “Toggle” component. In the Grasshopper workflow shown in Figure 5.1, the toggle output is connected to the “Simplify Lines” input parameter in the “MMesh” component. Here, the “Toggle” component allows the user to choose whether or not the “MMesh” component simplifies any line inputs or not; “True” means that lines are simplified, and “False” means that lines are not simplified.
5.2.10 The “Area Tolerance” Component¶
The “Area Tolerance” component specifies the minimum value of the ratio between an element’s area and the maximum area specified for the region of the mesh that the element belongs to. The “MMesh” routine attempts to create elements that are as close as possible to the maximum element area specified by the user. If the “MMesh” routine is not able to create elements that are large enough to meet the area tolerance criteria, it will disregard hard points so that it can. Therefore, if a curve has too many control points in one section, or if there is a cluster of wells too close together to allow for element sizes to meet the area tolerance criteria, the “MMesh” routine may disregard points that the user specified to match with mesh nodes.
5.3 Exporting a Completed Mesh¶
During the development of the mesh in Grasshopper, the mesh is shown as a preview that cannot be selected or modified in the Rhino workbook. Once the mesh is completed, the user may save the mesh in the Rhino workbook by right-clicking on the “MMesh” component and selecting the “Bake…” menu option, shown in Figure 5.11a. The user will be able to enter the name of the mesh and choose the layer it will be found in in the resulting menu, shown in Figure 5.11b.
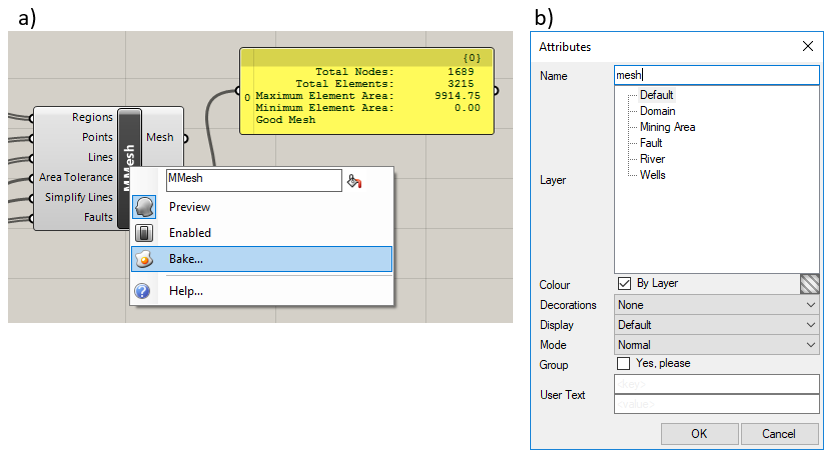
Figure 5.11 Saving a mesh: a) the “MMesh” component menu, b) the “Save Attributes” dialog box¶
In the Rhino workbook, the user can use the design tools to edit the mesh, if necessary. To export the mesh in a format compatible with MINEDW, first select the mesh, then choose “Export Selected…” in the “File” menu. Save the file as a stereolithography, or .STL, file.
5.4 The Rhino “Drop” Command¶
The “Drop” command in Rhino was developed to aid the creation of open-pit plans. The command allows the user to find the elevation of the pit at many x, y points without loss of accuracy due to interpolation. To install the function, go to the “File” menu and select the “Properties” option. The “Document Properties” dialog box shown in Figure 5.12 will open. Choose the option “Plug-ins” from the list on the left side of the “Document Properties” dialog box, circled in Figure 5.12. Click the “Install” button and choose the “Drop.rhp” file. Make sure that “Drop” is listed under “All Plug-ins” and that the “Enabled” box next to it is checked, as shown in Figure 5.12.
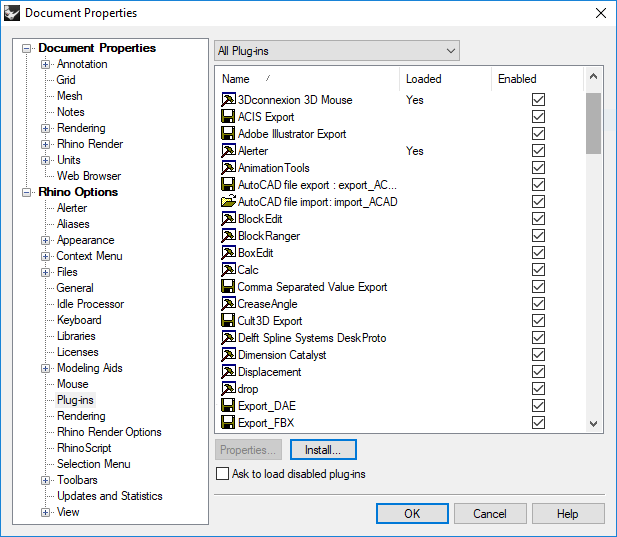
Figure 5.12 Installing the “Drop” command in Rhino¶
To use the function, open a mesh in Rhino. Import points to Rhino; the user may want the points to match the x, y locations of the model mesh’s nodes. Type “Drop” into the command window and, following the prompts, select the mesh, press [Enter], select the points, and press [Enter] again. The points will keep their x and y coordinates but change in elevation such that they are located on the mesh. The points may be selected and then exported from Rhino as a text file. The text file may then be imported into MINEDW for various uses.